

Mapping Drug Response Cascades with a Causal Gene Regulatory Network at 100M-Cell Scale
Learning a directed GRN from large-scale perturbations to explain how drug effects propagate through gene regulation.


INTRODUCTION
Learning Regulatory Cascades from Tahoe-100M
Gene regulatory networks provide a compact language for cellular behavior and response; which regulators appear to coordinate transcriptional programs, how those programs organize across cell types, and, when interventional evidence is available, how upstream changes propagate to downstream expression. In practice, most networks inferred from transcriptomes remain dominated by association. Single-cell RNA-seq amplifies this problem, strong latent state structure, cell cycle, stress, differentiation, together with technical effects, induces broad covariation, so many edges reflect shared context rather than direct regulation.
This is why “correlation is not causation” is not a slogan here; it is the identifiability problem.Two genes can rise and fall together because one regulates the other, because they share an upstream driver, or because both respond to an unmodeled shift in cellular state along a low-dimensional manifold. Motif priors and regulon-style approaches improve biological plausibility, but they do not, on their own, separate direct regulatory influence from downstream consequence. If we want directed edges that generalize, and not just descriptive modules, then we need information that is absent from observational snapshots. Perturbation datasets provide that information by injecting asymmetry into the data. With many interventions, especially when they span diverse targets, doses, and cellular backgrounds, the transcriptome becomes a readout of repeated “natural experiments,” where some changes are proximal and others reflect propagation through the network. Tahoe-100M was built to operate in that regime: ~100 million single-cell transcriptomes, spanning 379 compounds across 50 cell lines.
In this post we introduce TX1-CD (TX1 Causal Decoder), a structural-equation layer trained on top of Tahoe-X1(TX1) embeddings that decomposes each perturbation’s footprint into direct and cascading components, producing a directed GRN over 14,142 genes.
INFRASTRUCTURE
Eliminating Key Bottlenecks in Large-Scale Single-Cell Processing and Modeling with NVIDIA GPUs
Tahoe-100M pushes single-cell analysis into a regime where GPU acceleration isn’t optional. With ~ 100 million cells to process, even “standard” steps—normalization, covariate regression, neighbor graph construction, and large-scale gene–gene statistics—become throughput problems.
Our pipeline runs end-to-end on NVIDIA H200 GPUs. For single-cell preprocessing, we collaborated with scverse and NVIDIA to leverage rapids-singlecell (developed by scverse), which provides a drop-in replacement for Scanpy workflows on top of NVIDIA CUDA-X Data Science (DS). Under the hood, the core acceleration layer includes cuDF, cuML, cuGraph, cuVS, Dask-CUDA, cuBLAS, and RMM (RAPIDS Memory Manager); model training uses PyTorch DistributedDataParallel with the NCCL backend.
Pipeline Performance on 8× NVIDIA H200 (OCI Bare Metal)
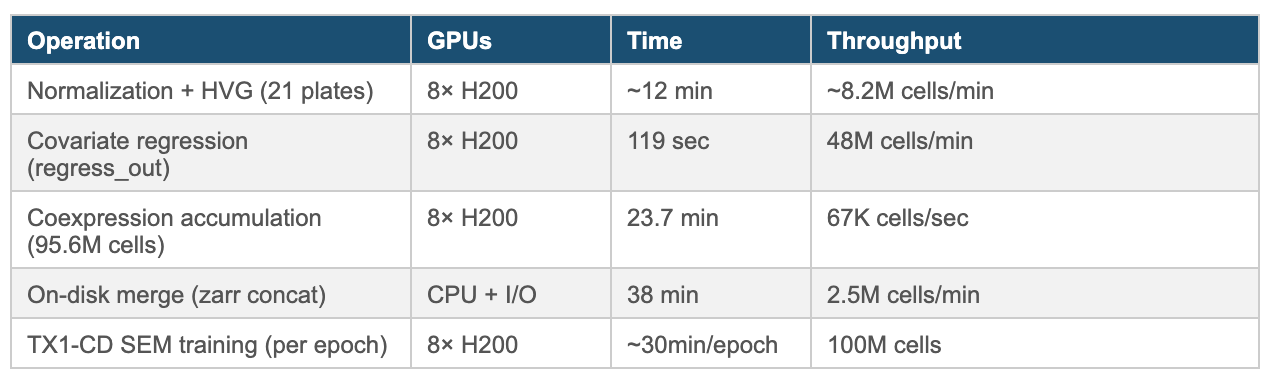
On a CPU-only cluster, this pipeline becomes impractical at Tahoe-100M scale: covariate regression across tens of millions of cells and dozens of covariates is both memory-intensive and slow, and large gene–gene coexpression accumulation quickly turns into a multi-node, multi-hour (often multi-day) exercise once you include end-to-end iteration and I/O. NVIDIA’s GPU ecosystem—CUDA-X DS-enabled single-cell preprocessing, cuBLAS-accelerated linear algebra, and PyTorch DDP with NCCL for training—compresses that workload into a single-node run where the compute-heavy stages complete in minutes and the full pipeline lands in roughly an hour on bare metal.
APPROACH
TX1-CD is a causal decoder trained on top of TX1 single-cell embeddings, designed to separate what a drug perturbs directly from what changes downstream through regulatory propagation. Tahoe-100M cells are first embedded with TX1 to obtain 2,560-dimensional per-cell representations, and we compute a gene-gene coexpression matrix across all ~100 million cells using a CUDA-X DS-based pipeline, Figure 1A. To keep inference tractable we sparsify this into a candidate-edge mask, we retain the top 100 coexpression partners per gene, we add curated transcription factor-target edges from CollecTRI, DoRothEA, and TRRUST, and we obtain roughly 1.16 million candidate edges, 0.58% density, over 14,142 genes, chosen to balance biological coverage and tractability, we take the union of curated regulator sets, transcription factors, RNA-binding proteins, cancer hallmark genes, and we augment them with the top 10,000 highly variable genes identified by per-plate consensus.
Training is two-stage, we first fit a basal decoder on DMSO cells to predict expression from cell context alone, then we freeze it to anchor the unperturbed baseline. We then introduce the causal module, each drug is represented by a sparse direct-effect vector dp, constrained to known target genes and their local neighborhoods, and we learn a directed GRN W under the candidate-edge mask with a low-rank parameterization. Drug effects are propagated through W with a truncated Neumann-series approximation, capturing multi-step cascades, and the final prediction is the frozen basal output plus the propagated drug component. We regularize W for interpretability and stability using edge dropout, delayed L1 sparsity weighted by prior confidence so curated edges are penalized less than novel ones, and spectral-radius clamping, ρ≤0.95, yielding a directed GRN over 14,142 genes.
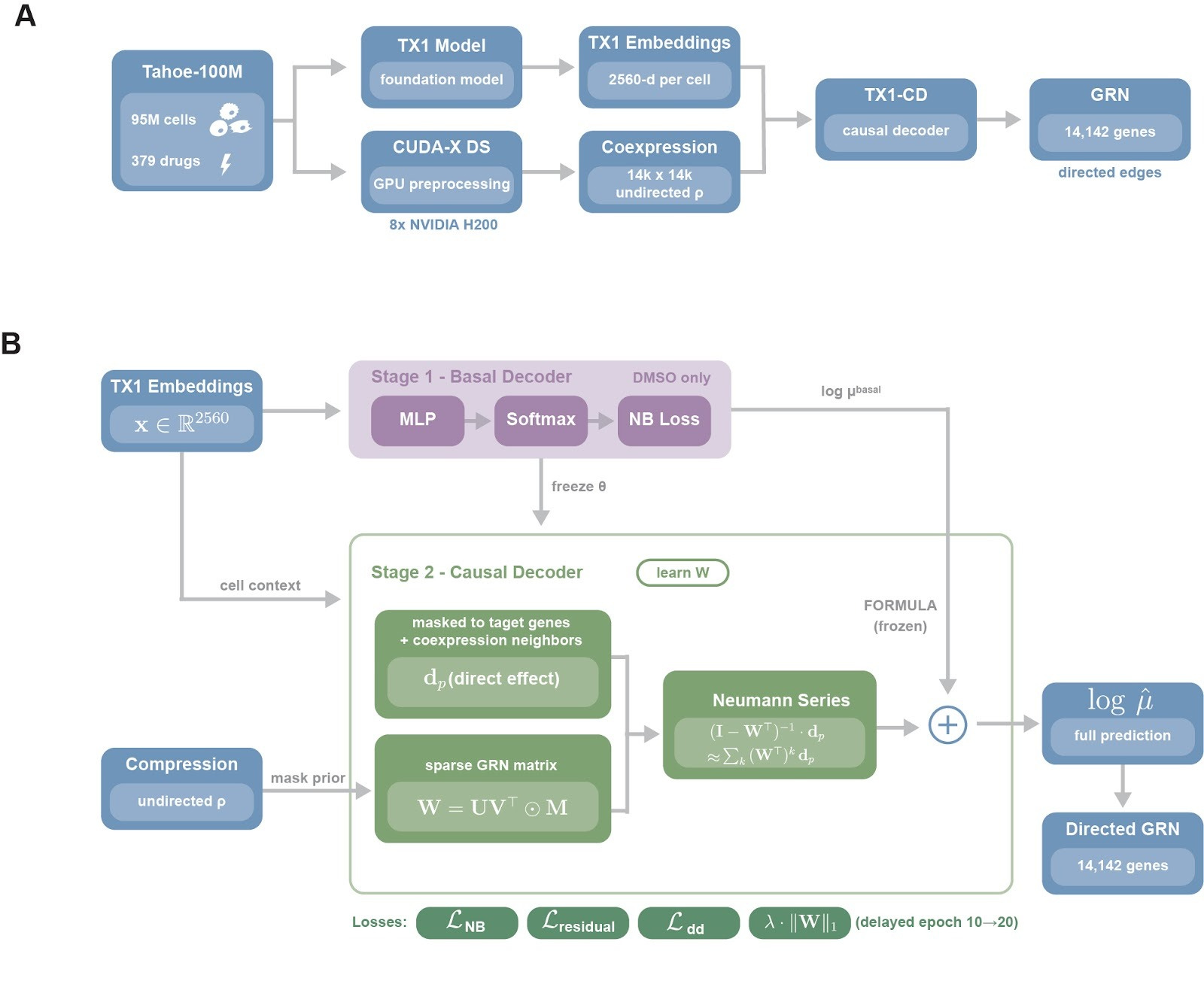
Tahoe-100M cells are embedded with TX1 and paired with a CUDA-X DS coexpression prior, TX1-CD is trained in two stages, a frozen basal decoder fit on DMSO cells, then a causal module that learns a sparse directed GRN and propagates drug direct effects through the network to model cascades.
KEY RESULT
The learned GRN captures biological structure that raw coexpression does not
Our expectation is that the learned directed GRN captures structure beyond generic coexpression. To test this assertion, we evaluated the learned weights of GRN against four independent external references, STRING physical interactions, EBI Complex Portal and hu.MAP2 protein complexes, and Reactome pathway co-membership, scoring GRN and raw coexpression matrix on the exact same ~1.16M candidate edges defined by our mask. In this controlled hypothesis space, coexpression provides limited ranking signal, whereas TX1-CD consistently prioritizes edges supported by these orthogonal functional benchmarks, improving AUROC from 0.42–0.58 to 0.74–0.83, Fig. 2A. The learned network also exhibits the expected directionality, known transcription factors are enriched among the top out-degree hubs, and annotated drug targets are enriched among the top in-degree nodes, Fig. 2B. Together, these results suggest TX1-CD is not merely reweighting correlation, it learns a directed regulatory structure that aligns with known biology and places regulators and targets into distinct topological roles.
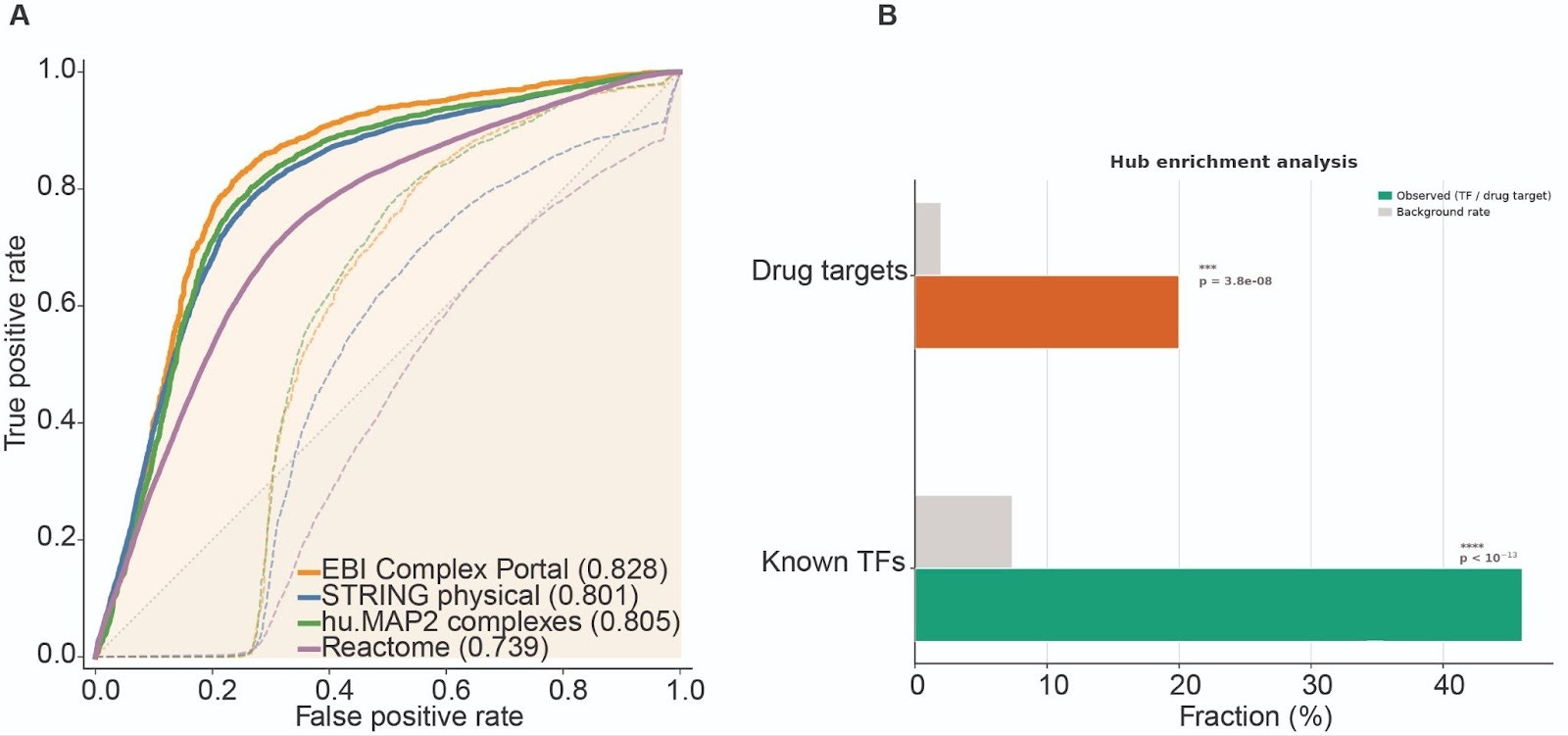
(A) ROC curves comparing edge rankings from raw coexpression and learned weights GRN against four independent references, evaluated on the same ~1.16M candidate edges. (B) Hub enrichment showing over-representation of known transcription factors among top out-degree nodes and annotated drug targets among top in-degree nodes.
OUTLOOK
What Comes Next
As we continue to exponentially grow Tahoe’s datasets with new perturbations and cellular contexts, we will scale TX1-CD training accordingly, longer runs on larger, more diverse interventional corpora are the most direct way to sharpen the separation between direct effects and downstream cascades, and to expand the regulatory resolution the model can support. To make that scaling practical, we are moving more of the workflow onto next-generation NVIDIA GPUs, including B200, and pushing both preprocessing and model development into a fully GPU-native, multi-node regime. On the preprocessing side this means deeper adoption of CUDA-X Data Science libraries, as well as rapids-singlecell, and on the modeling side it means scaling training efficiently through distributed sharding, for example nvFSDP-style approaches, so that throughput increases without host-device movement or memory fragmentation becoming the limiting constraint.
With that foundation, the goal is not simply a larger network, it is a more useful one for mechanism. TX1-CD learns a directed topology with the expected roles, high out-degree hubs are enriched for transcription factors, 46% of the top 50, p = 10⁻²⁸, while high in-degree nodes are enriched for known drug targets, 20%, p = 10⁻¹⁰, and these hub identities emerge from perturbation-driven signal propagation rather than being hard-coded by priors. More importantly, for each compound the model decomposes the response into a proximal signature and a propagated cascade, which lets us trace putative mechanism as paths through regulators and bottlenecks, prioritize drug combinations when distinct compounds converge on shared downstream control nodes, and flag high-confidence edges missing from existing references as testable hypotheses. As the GRN expands, it can be directly overlaid with curated gene sets and regulatory modules (e.g., Gene Ontology), revealing regulatory connections around canonical pathways such as MAPK or JAK–STAT that are invisible in traditional pathway maps. These connections point to new nodes and targets for therapeutic intervention.
Finally, models like TX1 and TX1-CD are intended to be foundations, not endpoints. Our vision is that autonomous research agents can reason on top of them to simulate and stress-test mechanistic hypotheses about drug response, tracing candidate cascades, comparing them across contexts, and proposing focused follow-up perturbations that would most clearly validate or falsify the explanation.
Built with PyTorch · NVIDIA CUDA-X / BioNeMo Platform · rapids-singlecell (scverse) · CuPy (Preferred Networks) · NCCL · Dask-CUDA · RMM
TX1 Foundation Model · Tahoe-100M Atlas
Wow! Is this data going to be released on huggingface?